
If you have worked in European regulatory affairs for some time, it’s hard to wrap your brain around the fact that the United Kingdom is no longer part of the European Union. The UK will once again forge its own path, interacting with its continental neighbors in a manner similar to Norway. Yet the key difference is that Norway has chosen to emulate the EU Medical Device Regulation (2017/745) as national law and require CE Marking.
In late 2020 the UK MHRA further ensured job security among medical device regulatory professionals by moving to implement their own registration and marking scheme.
An understanding of the new UK MHRA medical device registration requirements starts with a basic UK geography lesson. Brexit created many logistical and political headaches, one of which is what to do about cross-border trade between Northern Ireland (part of the UK) and the country of Ireland (an EU member). As such, a scheme was created that treats Northern Ireland and Great Britain (England, Wales, Scotland) differently from a regulatory perspective, even though all four regions are part of the UK. This is the side effect of an effort to prevent a “hard border” between Ireland and Northern Ireland.

Now, device manufacturers selling in the UK must follow the UK Medicines and Medical Devices Act of 2021 (MMDA). Valid CE Marking certificates issued by EU Notified Bodies will continue to be recognized in the UK until June 30, 2023.
![]()
Medical device manufacturers must apply a new UK Conformity Assessed (UKCA) Mark to their device labeling for products sold in England, Wales, and Scotland. This means that if you have a current, valid Declaration of Conformity or CE Marking certificate, you automatically meet UKCA mark requirements. However, you will not need to modify your existing labeling to add the UKCA mark until July 1, 2023. After this date UKCA it will be mandatory, but dual labels with the CE and UKCA Marks will be acceptable.
As for new devices, a UK Conformity Assessment Body will need to a conduct an assessment for compliance with the UKCA mark requirements. Existing UK Notified Bodies under the European MDD most likely will continue to be designated for UKCA conformity assessment according to CE Marking. However, note the conformity assessment will be against the UK medical device regulations, which follow the MDD.

As if Brexit wasn’t confusing enough, the UKCA mark will not be recognized in Northern Ireland which will have its own UKNI mark! If you want to make your head hurt even more than it already does, you can read about that know more. You better design larger labels to account for all these new marks!
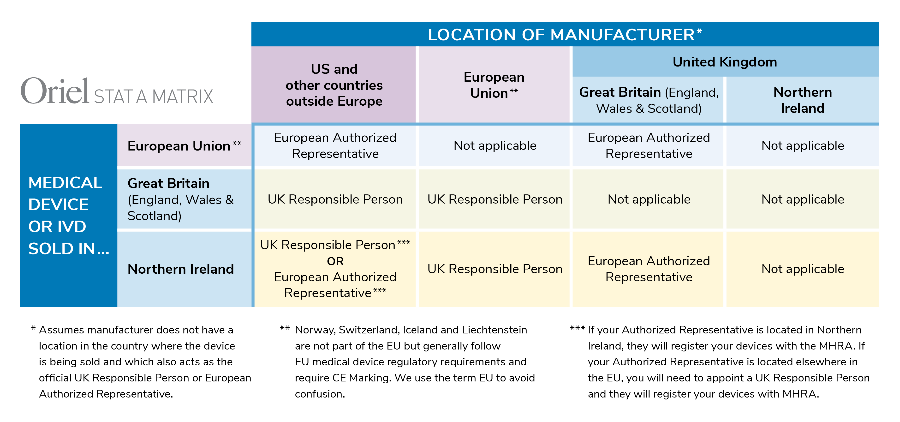
In addition to the new UKCA mark, manufacturers located outside of the UK will need to appoint a UK Responsible Person prior to registration. This applies to products sold in England, Scotland and Wales (collectively Great Britain). Similar to the EU Authorized Representative, the UK Responsible Person will be responsible for registering devices with the MHRA, ensuring that a Declaration of Conformity has been drawn up and that technical documentation exists. Various other responsibilities apply as well.
For devices sold in Northern Ireland, things get really tricky. If the manufacturer is located in Great Britain, an EU Authorized Representative is required, not a UK Responsible Person. If the company is located outside the UK, a UK Responsible Person will be required, along with a UK importer.
The process of untangling nearly 50 years of political, trade, and legal integration with the EU is messy and ongoing. There is plenty more to do, and the MHRA is surely working hard to establish a new framework for regulation of medical devices in the UK. As things evolve, Oriel STAT A MATRIX will keep you updated.

US OfficeWashington DC
EU OfficeCork, Ireland


