
The EU Medical Device Regulation (MDR 2017/745) and EU In Vitro Diagnostic Regulation (IVDR 2017/746) put forward a multitude of challenges, chief among them the process of clinical evaluation. For medical device and IVD companies making products that incorporate software, the question of how much clinical evidence to gather can be perplexing.
Rest assured that you are not the only one scratching your head. Many are apprehensive about how Notified Bodies will evaluate clinical evaluation reports (CER) or IVD performance evaluation reports (PER) for devices incorporating software or as stand-alone software.
Shortly after the release of the EU MDR and IVDR, many device manufacturers with products that include software began to question clinical evidence requirements: Do I need to prepare a CER or PER for the device and the software separately or as a system? How do I show clinical evidence for a device that now incorporates software? What do I assemble for stand-alone software with clinical decision parameters?
The challenge is that software can be used alone or in combination with a device sometimes driving its operation, sometimes used as stand-alone application, or sometimes used as an accessory. This can make the decision on how to approach clinical requirements tricky.
One thing is crystal clear: Any software that qualifies as a medical device or IVD must meet all clinical evaluation and performance evaluation principles outlined in Article 61(1) of the MDR and Article 56(1) of the IVDR.
The Articles state: The manufacturer shall specify and justify the level of the clinical evidence necessary to demonstrate conformity with the relevant general safety and performance requirements. That level of clinical evidence shall be appropriate in view of the characteristics of the device and its intended purpose.
As such, manufacturers of medical device software must:
However, as we noted earlier, it’s not always clear whether medical device software should be evaluated with the medical device or separately. To add some clarity to the issue, the Medical Device Coordination Group issued guidance MDCG 2020-1.
The core issue comes down to the role software plays in your device and whether you need to conduct a CER or PER on the software itself or as a system with the connected medical device or IVD. This will help you determine how to approach your clinical evaluation or performance evaluation, and subsequently keep you out of hot water with your Notified Body during your product certification evaluation. Generally, any software integrated directly into a medical device or IVD should be treated as a system. Annex II of MDCG 2020-1 provides some helpful examples that can assist you in determining whether your evaluation should be done on the device as a system, device, and software separately or just on the software.
Stand-alone software that takes CT scan data from an already performed diagnosis and completes an algorithm calculation for each slice of CT data to identify potential regions of interest for a radiologist to investigate further for potential cancer identification.
IVD software application that takes existing data from multiple results of IVD biomarker tests and, using an algorithm, compiles biomarker results and compares against multiple databases to provide analysis of potential genetic disorders.
The software application resides on the hospital server connected to three other medical devices where central monitoring can occur by healthcare professionals and the software can turn on or off functions for each device remotely by a healthcare professional.
The software is controlling the insulin pump by connecting to it. However, the software does not have any medical purpose on its own. As such, the clinical evaluation should be performed together with the pump and not independently.
Because this software is part of a closed-loop system, the clinical evaluation should not be limited to the software and should include preclinical and clinical investigations for the entire system.
Section 4 of MCG 2020-1 offers some details on how to think about your clinical evidence strategy for software. Annex I on page 17 of the guidance offers a useful decision tree that can guide your generation of clinical evidence for software.
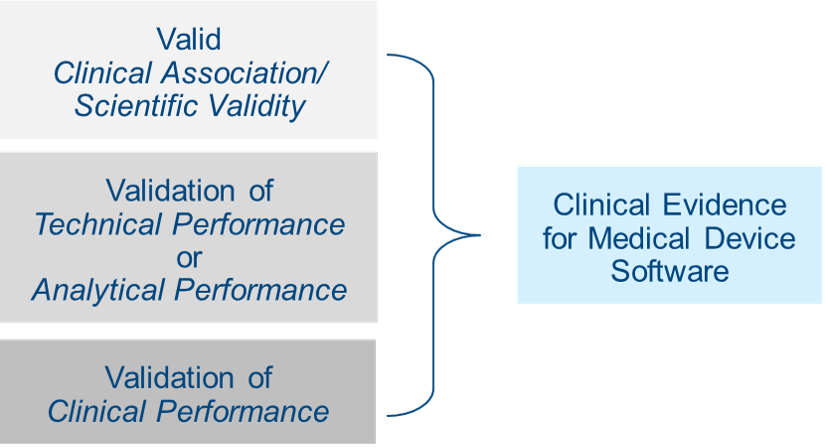
According to Section 4.1 of MDCG 2020-1, this means the extent to which the (software) output is based on the inputs and algorithms selected, is associated with the targeted physiological state or clinical condition. This association should be well founded OR clinically accepted. This means that it must be generally accepted by the medical community and/or described in peer-reviewed scientific literature. Section 4.2 notes that evidence supporting this can be gathered through literature research, professional guidelines, proof-of-concept studies, or your own clinical investigations or clinical performance studies. Existing data can include technical standards, literature reviews, published clinical data, or your own clinical investigations/clinical performance studies. Be mindful that software can include many clinical features that require individual assessment, and that current state of the art should always be taken into account. Read what this means.
The goal here is to make sure your software reliably, accurately, and consistently meets your stated intended purpose in a real-world setting. Performance validation and verification can be demonstrated through various clinical and performance perspectives of data, including availability, confidentiality, integrity, reliability, accuracy, analytical sensitivity, limit of detection, limit of quantitation, analytical specificity, linearity, cut-off value(s), measuring interval (range), generalizability, expected data rate or quality, absence of acceptable cybersecurity vulnerabilities, and human factors engineering.
The guidance recommends that evidence supporting performance be generated through verification and validation activities at the software unit level, integration, system testing, or by generating new evidence using curated databases, registries, reference databases, or previously collected patient data. See section 4.3 of the guidance document for more information on this.
Does software generate clinically relevant output in accordance with the intended purpose? Does the software output result in a positive impact on the health, diagnosis, or treatment of the patient, or on patient management? The best way to gather this evidence is testing the medical device software for its intended use among the target population and under typical use conditions. Get more details on this Know More.
It is important that you consider validation of clinical performance at each release of software. If you choose not to perform validation, then you need to justify why in your technical documentation.
What if the device has multiple features but only some of them offer clinical benefits? If that’s the case, then validation of clinical performance may only be applicable to those features. This brings up another point: Sometimes software can be modular. If that’s the case for your product, then it is permissible to validate clinical performance for each module as long as its functionality is independent of the other modules. The benefit of this approach is that it allows for continuous risk acceptability for each module that has changed.
What if the software application or feature has no clinical benefits? If so, clinically relevant outputs can be demonstrated through predictability, reliability, and usability. Of course, there is far more involved in validating clinical performance of medical device software than we can outline in this article, so please be sure to read section 4.4 of the guidance document carefully.
The issue of clinical evidence and how to approach it obviously dovetails into your overall risk management strategy. As such, its worth taking a step back and reading this article on risk management for medical device software and SaMD.
If you made it all the way to the end of this article, you’re obviously committed to learning as much as possible about clinical evidence for software. You may still have questions and we can help in several ways. Our MDR/IVDR consulting team is available to assist with EU MDR/IVDR gap assessments or clinical evaluation report development/review. Or consider taking your knowledge to the next level by registering for our Medical Device Software Development, Verification, and Validation Training class, or our Medical Device Risk Management Training class. Both are available as virtual training or in-person formats.

US OfficeWashington DC
EU OfficeCork, Ireland


