QA/RA Consulting, Auditing & Training

The one thing that remains constant in the medical device world is the frequency and need for change. In fact, the emphasis on the “management” of change has increased over the past few years with changes in the regulations and standards. It is the key process that drives our ability to manage those ongoing activities that impact our business and quality management system. Without a change management process, it would be easy to lose control of design and manufacturing activities.
The FDA and other regulatory agencies have long mandated that medical device manufacturers maintain a formal and systematic approach to manage and control changes to products, processes, procedures, equipment and systems. Additionally, change management activities have been integrated into key processes such as CAPA, complaint management post market surveillance, and risk management. Overall, management of change drives the expectation that manufacturers will continually improve their products and processes while ensuring necessary changes are implemented and managed in a controlled manner.
Whether you are dealing with transition to the EU MDR or IVDR, addressing compliance to FDA or other country requirements, or conformance to ISO 13485 and 14971, it is critical to establish a strong change management program and to evaluate ANY changes within the operation. This evaluation identifies significant changes that may impact your regulatory requirements or product registration requirements. While individual countries may have different definitions and requirements for the handling of significant changes, they are generally similar. All country regulations require management of change starting in the design phases of the product throughout the full life cycle of the product through the post market surveillance. Your management of change process should be implemented to ensure any change is thoroughly evaluated and does not introduce unintended negative consequences or modifications in products or processes. It is not designed to be just a paperwork exercise. A breakdown in the management of change process can result in product recalls, regulatory penalties, negative publicity, or harm to patients.
Change management begins once the design inputs and outputs have been aligned and are relatively stable. As the product moves through the design process to commercialization, management of change ensures the design and the appropriate documentation stay aligned and consistent with the defined regulatory strategy and documentation. Remember, the FDA requires inputs must equal outputs. The management of change process help to achieve this goal. Additionally, changes are routinely evaluated for impact on the risk management documentation:
The design development plan should identify the appropriate stages for the review and management of changes.
Training is a critical part of the overall communication plan. Ensure all parties involved have been informed and trained on the change and any impact it may have on their job.
The key activities in the product life cycle that require management of change include:
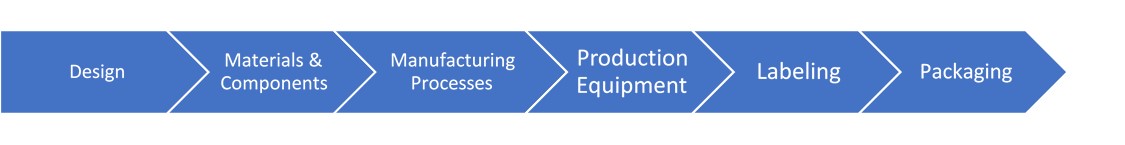
Each change must be evaluated individually as well as in aggregate to other changes for potential impact on the design of the product or process A minor change by itself may have no impact on other parts of the QMS, product lifecycle, or regulatory strategy. When you look at multiple minor changes in aggregate, they may add up to a major or critical change in the eyes of the regulator. As such, you might need to update submissions for approvals or modify your certifications.
Once the change request form is received, it needs to be reviewed (step 3) on its merits. If the change seems appropriate, a thorough assessment of all possible effects and risks needs to be conducted. You’ll also need to determine if any special equipment or personnel is needed to make the change. At this point you should have a procedure in place to broadly categorize proposed changes. Many companies use a simple three tier system.
Remember to include ALL changes for the evaluation, even those from the corporate office or outside the facility. Suppliers have been known to make changes in materials or processes that may have some impact on your product. Ensure your process includes how these are managed.

By its very nature, a change management system covers all changes whether they were planned or not. Anything that is unplanned is considered a deviation or nonconformity and must be addressed. All employees must be trained to distinguish between a deviation (unplanned) and a planned change. The CAPA system is often used to address the root cause of any unplanned change or deviation to determine what happened. Also, risks to the product or process need to be evaluated.
With regard to planned changes, it is important that you carefully document steps previously described and have the appropriate management review and approve (step 4) any changes before making them. It’s very easy to slip into a bad habit of “just making a quick change” in an effort to be nimble but those shortcuts may come back to bite you when an FDA inspector or Notified Body asks to see the documentation. Management of change documentation must be filed for easy access and review as it is considered part of the design history file, the risk management files, as well as post market surveillance. Keep your process simple and there is greater probability of conformance.
Your management of change procedure must specify which groups will be involved in reviewing a proposed change and the responsibility of each group. Typically, the regulatory, QA and facilities departments are involved, and possibly staff from R&D/engineering, safety, and environmental. Your change management record must include a description of the proposed change, identify affected documents, have the signature of the appropriate management person, an approval date and when the change goes into effect. It should also include procedures for updating primary and secondary documentation. This “secondary” documentation (e.g., component drawings, risk management files, IFU, and packaging) often gets forgotten.
All that being said, it’s easy to get carried away when creating a procedure. Your goal should be to create a procedure that’s easy to understand, clearly defines the levels of justification and approval needed, and the specific responsibilities of the people responsible for approval. You should also Include provisions for handling emergency changes when speed is of essence. Ultimately the design of your management of change system will be a function of the type and complexity of your products and operations, plus size and structure of the company.
In general, proposed changes should be encouraged but vetted with extreme care. You want employees to suggest changes in the name of continuous improvement but clearly not all change requests offer enough benefits to offset the risks. Rather than handling every change in an ad hoc manner, consider implementation of a two-tiered approach with escalation High priority or emergency types of changes are evaluated immediately just like potentially reportable incidents. Routine management of change requests could be reviewed monthly so people with requests can submit their proposals. It would be best if all people responsible for approving requests were present along with representatives from QA & RA. Provide all supporting materials in advance so appropriate people can think about the proposal and have a chance to ask relevant questions during the meeting.
Training is also essential to the success of any management of change system. Employees and managers must know how the management of change system and be willing to use it. With regard to the latter, this means having management of change request forms that don’t look like mortgage applications. All employees should be able to initiate changes and giving employees opportunities to practice is far better than providing remedial training after problems occur.
The need for robust management of change is outlined in three specific subparts of FDA 21 CFR Part 820 (aka, Quality System Regulation) and throughout ISO 13485:2016. The regulation and standard require that manufacturers establish written management of change procedures that describe company approved procedures.
US FDA QSR (21 CFR Part 820)
ISO 13485:2016
MDCG 2020-3 Rev 1
There’s a lot to know about management of change best practices and we have only scratched the surface. If you would like to learn more best practices for process mapping and writing standard operating procedures, consider this training course. Our risk management and PMS courses are also (incomplete sentence)

US OfficeWashington DC
EU OfficeCork, Ireland



UNITED STATES
1055 Thomas Jefferson St. NW
Suite 304
Washington, DC 20007
Phone: 1.800.472.6477
EUROPE
4 Emmet House, Barrack Square
Ballincollig
Cork, Ireland
Phone: +353 21 212 8530